深势·宇知
玻尔空间站

生命科学
物质科学
勒贝格智算
了解我们
EN


目标愿景
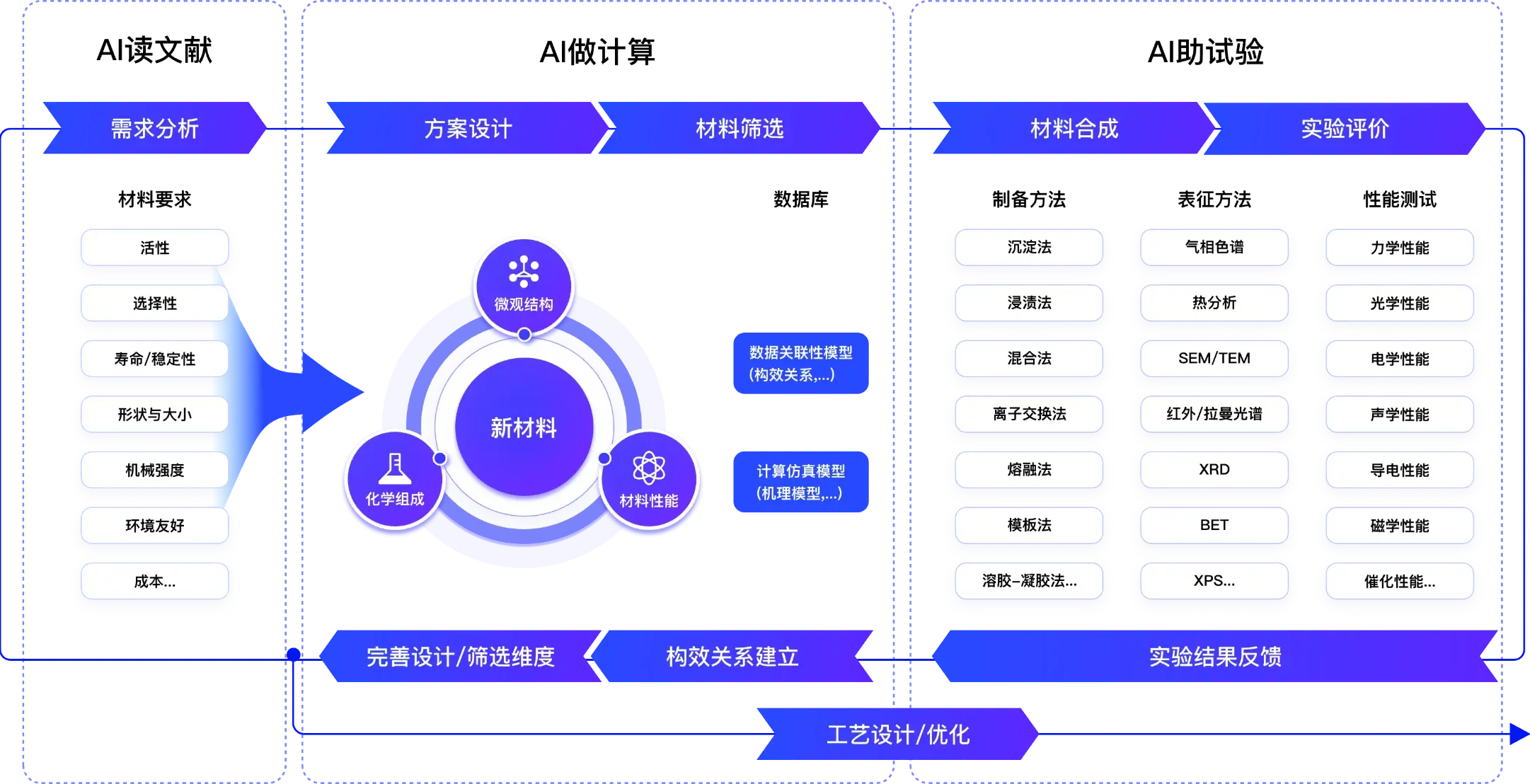
提升新材料研发效能




应用场景
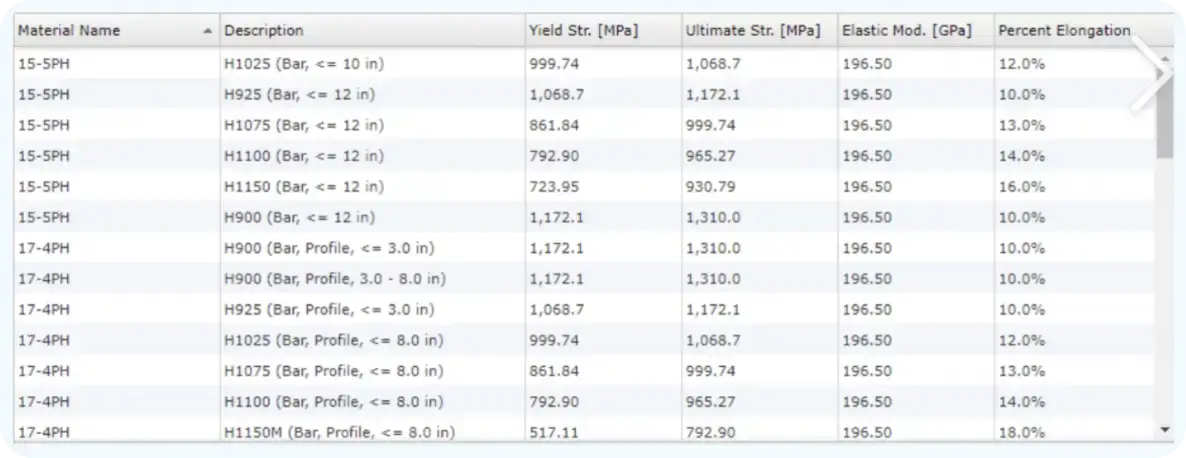
合金材料
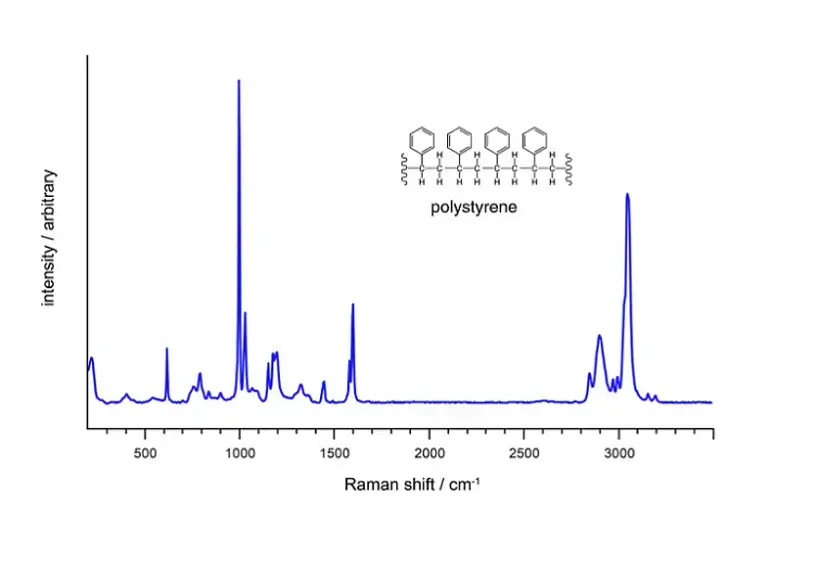
聚合物材料
催化材料
光电材料
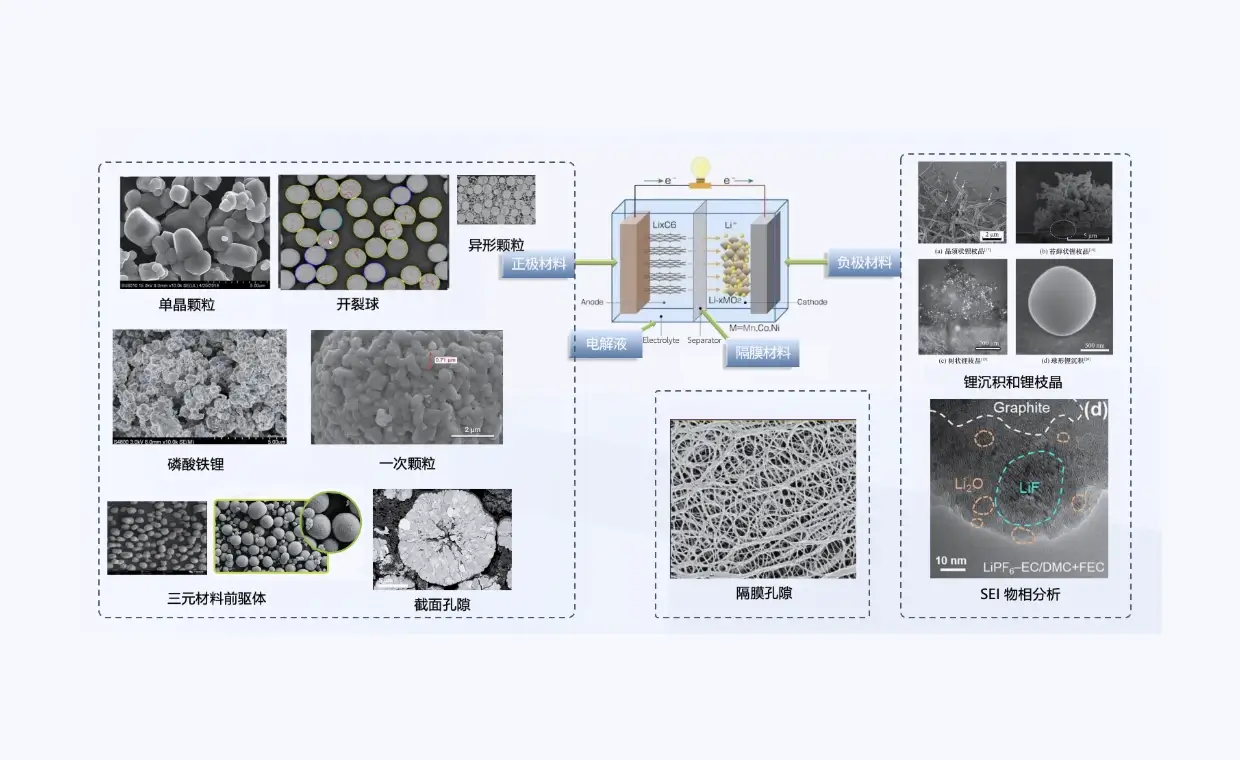
电池材料

Preview
融合云原生高性能计算引擎与先进机器学习模型,推动新型合金材料成分设计
高通量预测弹性模量、缺陷形成能等多种合金属性
快速评估合金材料的稳定性
研发痛点
1
巨大的搜索空间


2
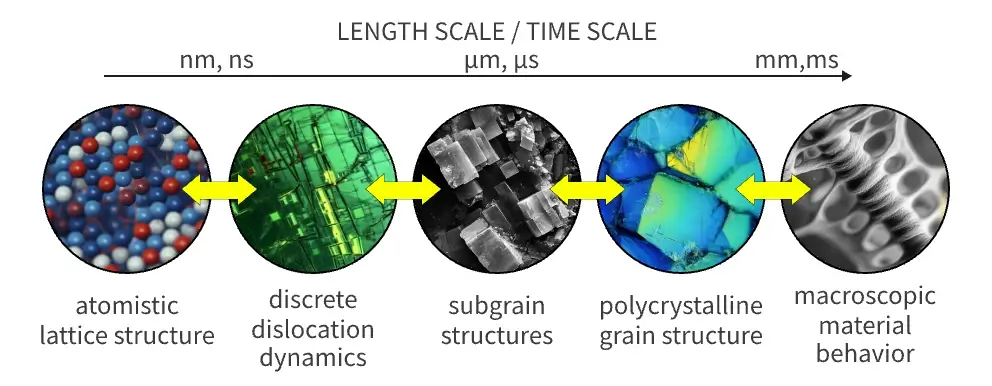
缺少跨尺度理解
3
复杂数据管理

软件功能
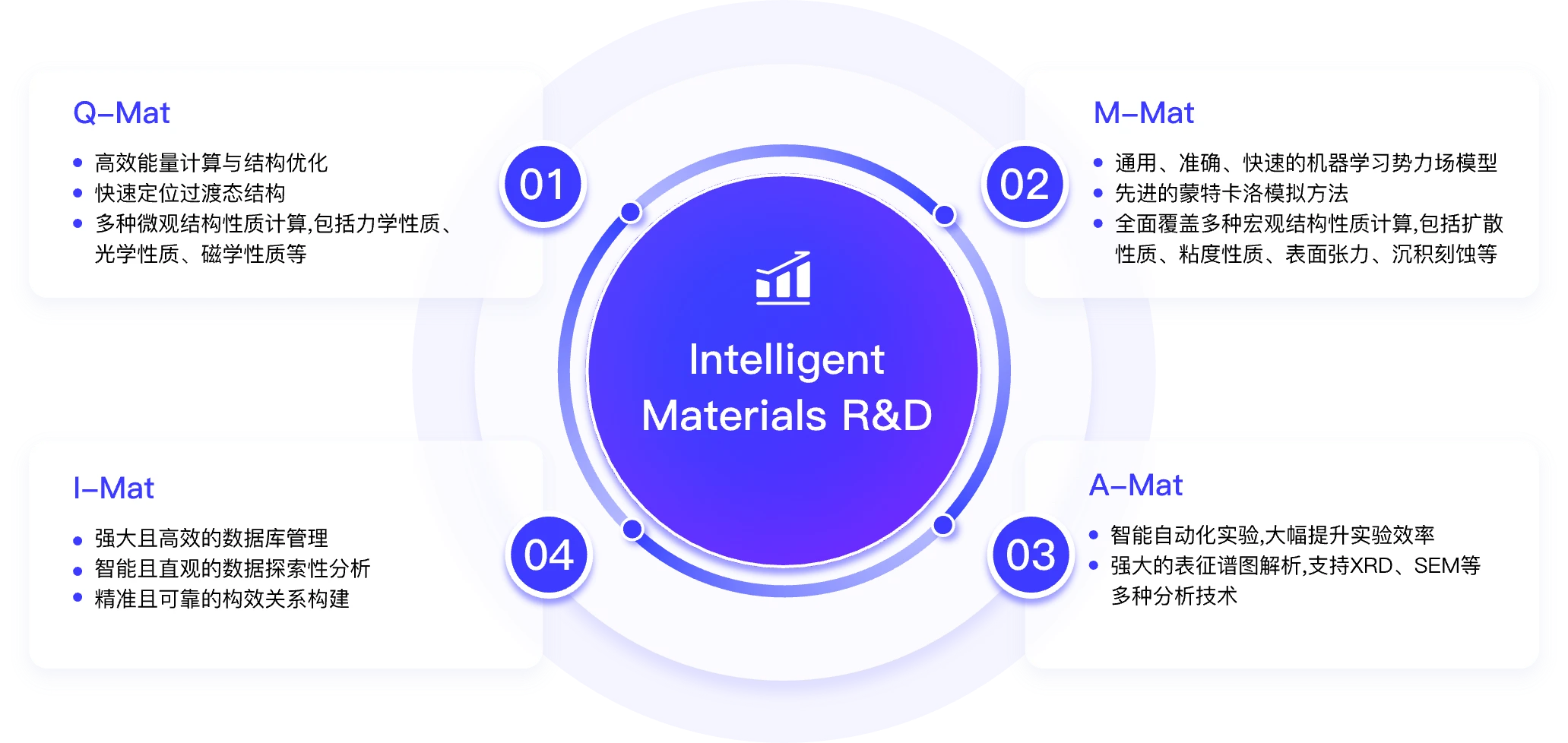
研发服务
实践案例
Preview
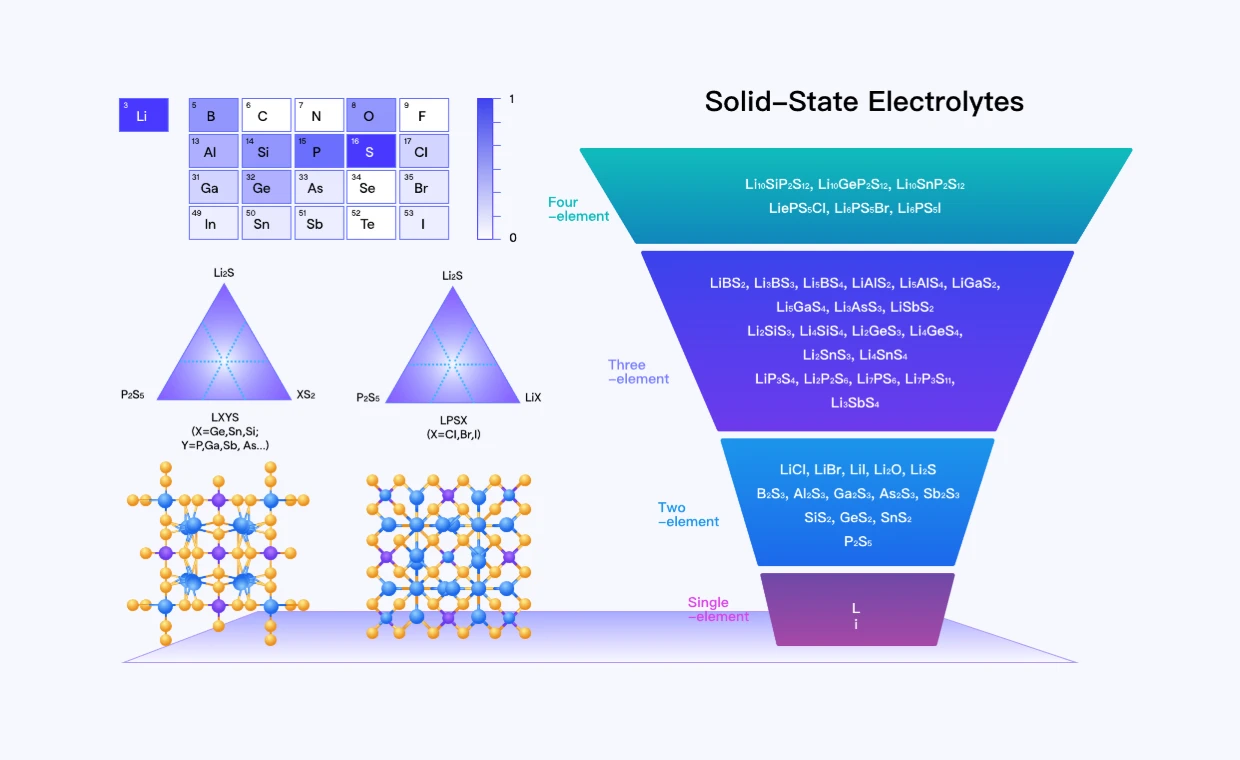
固态电池材料设计与改性
通过固态电解质原子仿真大模型准确预测固态电解质离子电导与稳定性,覆盖超过27种元素,辅助实验高通量筛选潜在候选固态电解质材料。

电池材料

Preview
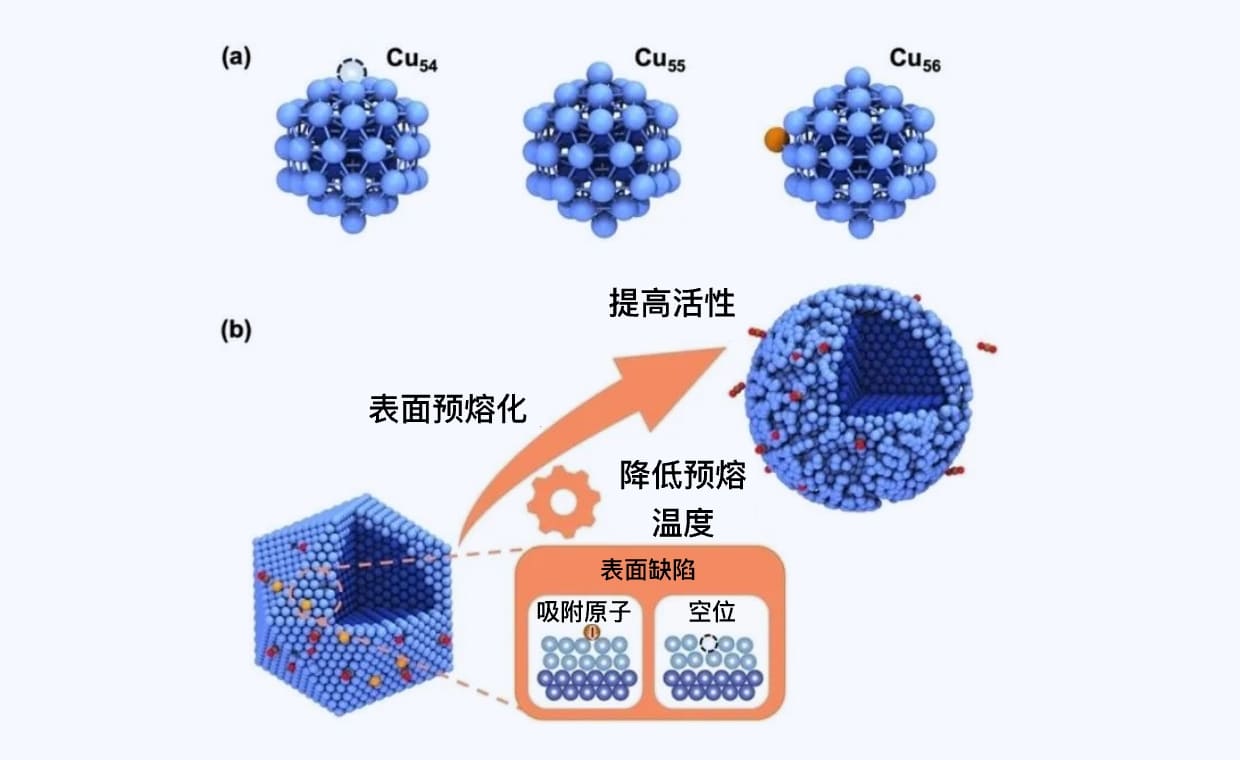
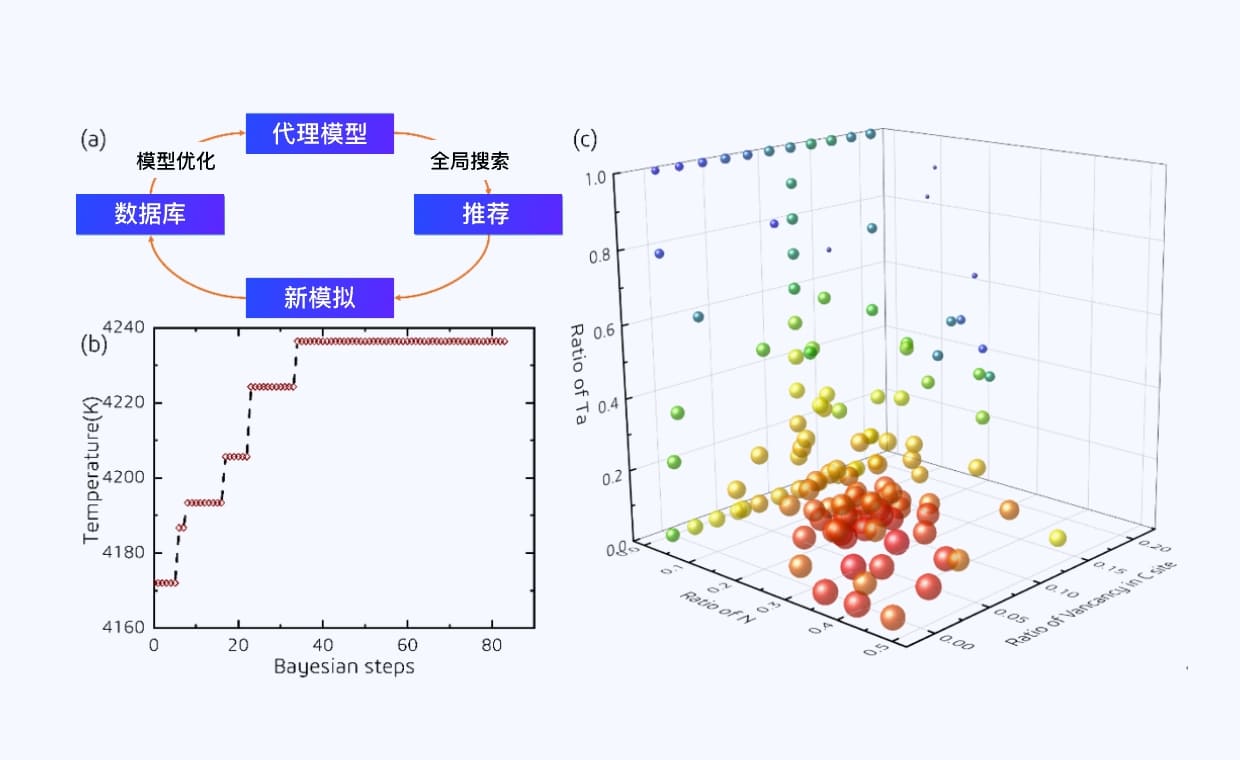
数字化复现高温哈伯法中催化反应的真实过程
基于深度势能分子动力学与动态概率增强采样方法,突破模拟的空间和时间尺度限制,理解高温条件下催化剂表面的真实状态,以及催化反应和催化剂毒化失活机制。
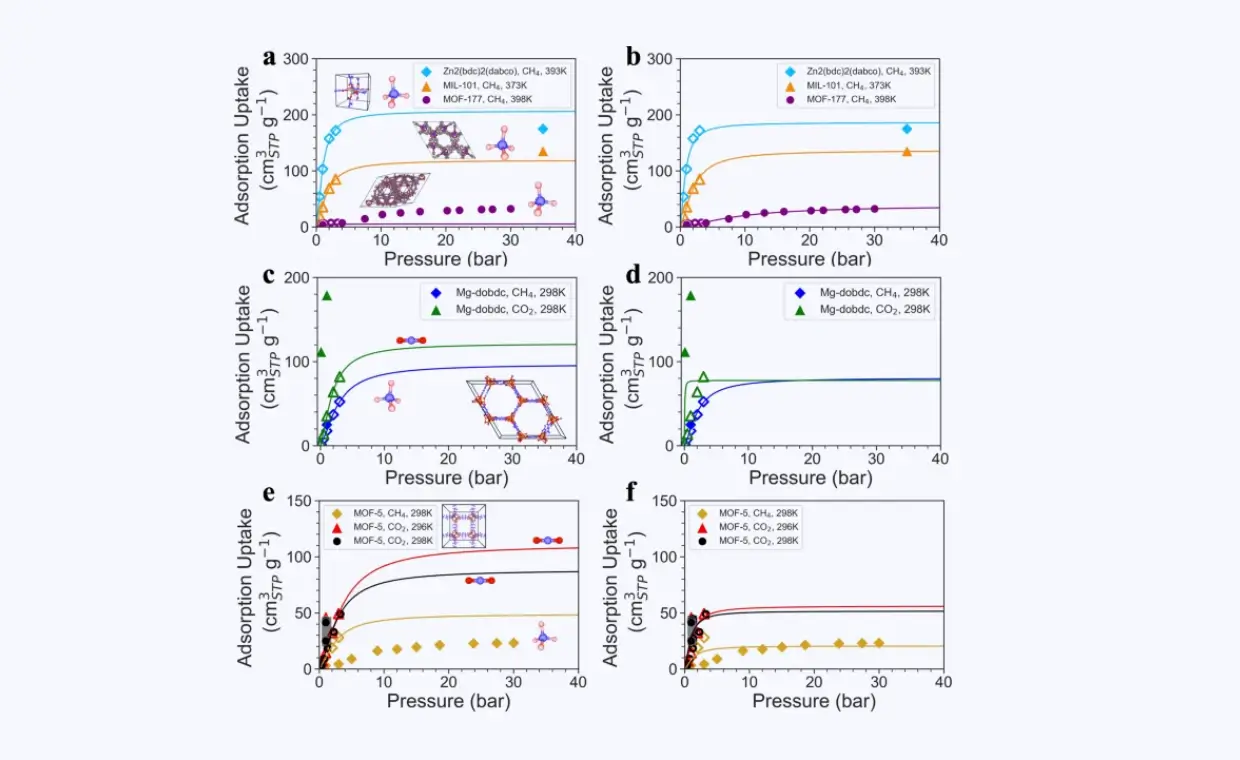
催化材料
Preview
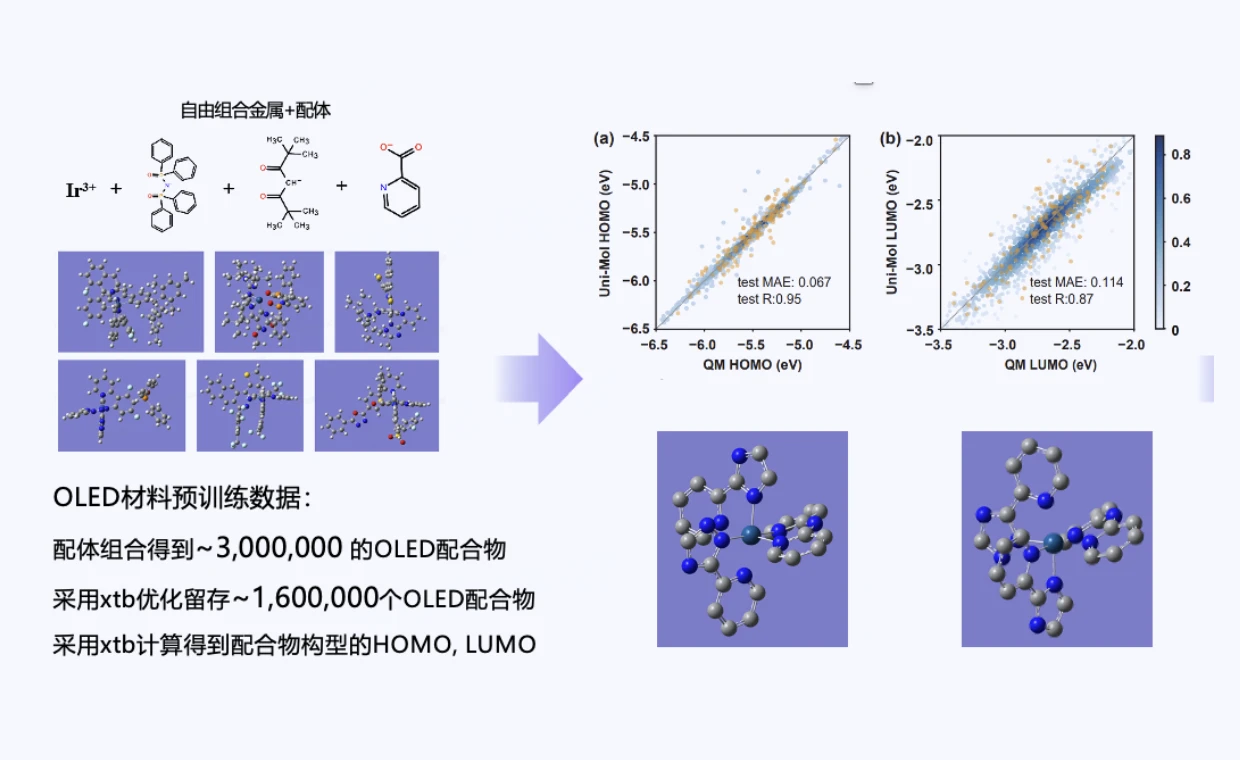
二代OLED分子Ir(III)配合物的高效筛选
通用分子设基座模型Uni-Mol实现了仅依赖极少量的量子力学计算数据建立可靠的结构-性质关系,并通过随机组合Ir(III)和有机配体的方式在量子化学精度下得到了百万级的潜在有机配合物数据库,并发现了具备优良光学性质的潜在专利分子。
功能材料
合作伙伴








