
Hermite®
AI4S-Enhanced CADD Tool for Accelerating Rational Drug Discovery
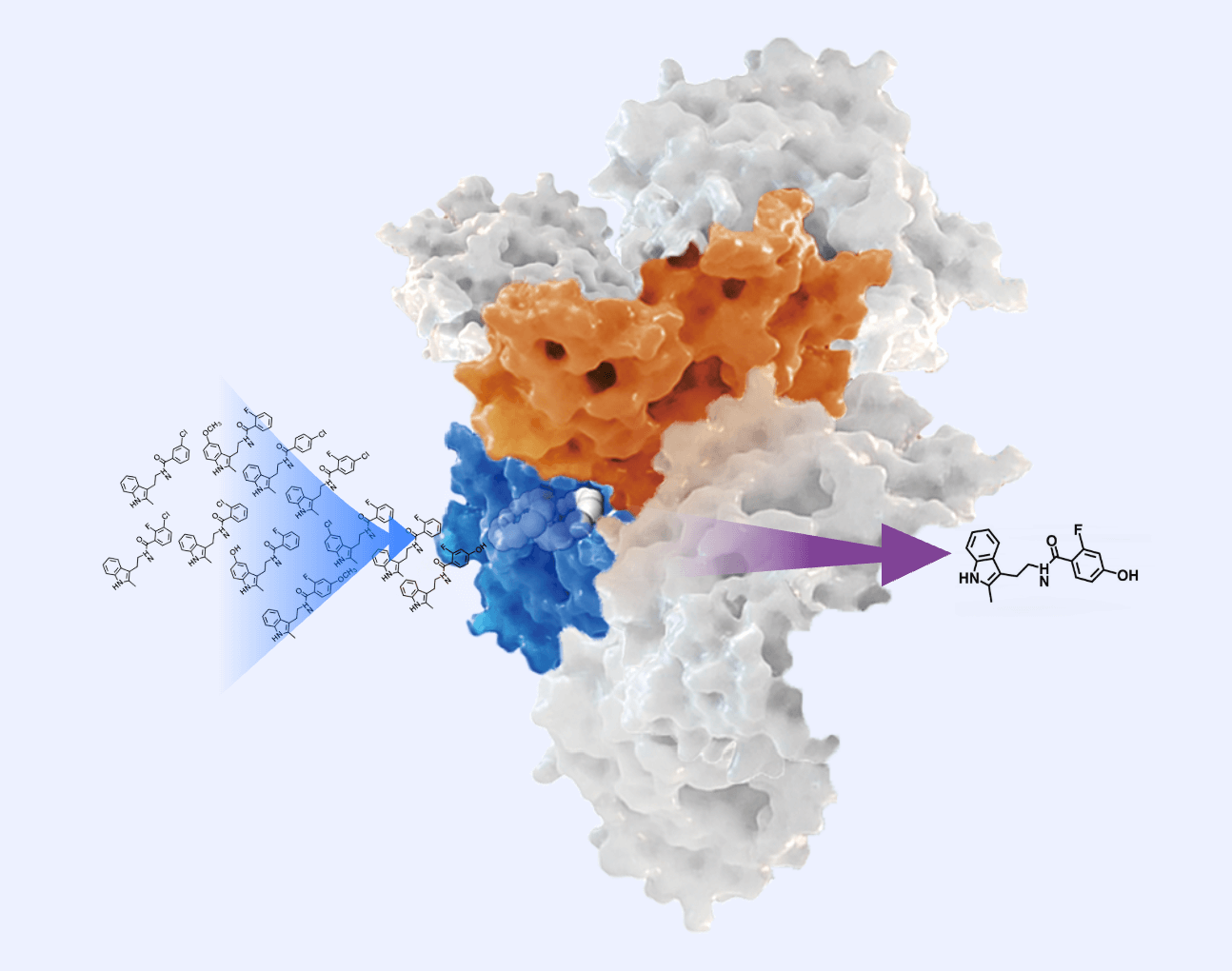
Hermite® combines artificial intelligence, physical modeling, and high-performance computing to streamline preclinical drug discovery.
Hermite®, with massive cloud-based computational resources, supports large-scale hit compound screening and lead compound optimization, all within a single platform. Hermite® web-based molecular visualization tools enable seamless collaboration and data sharing, allowing researchers to view, design, and analyze molecular data across devices.




Custom Visualization Component Library for CADD
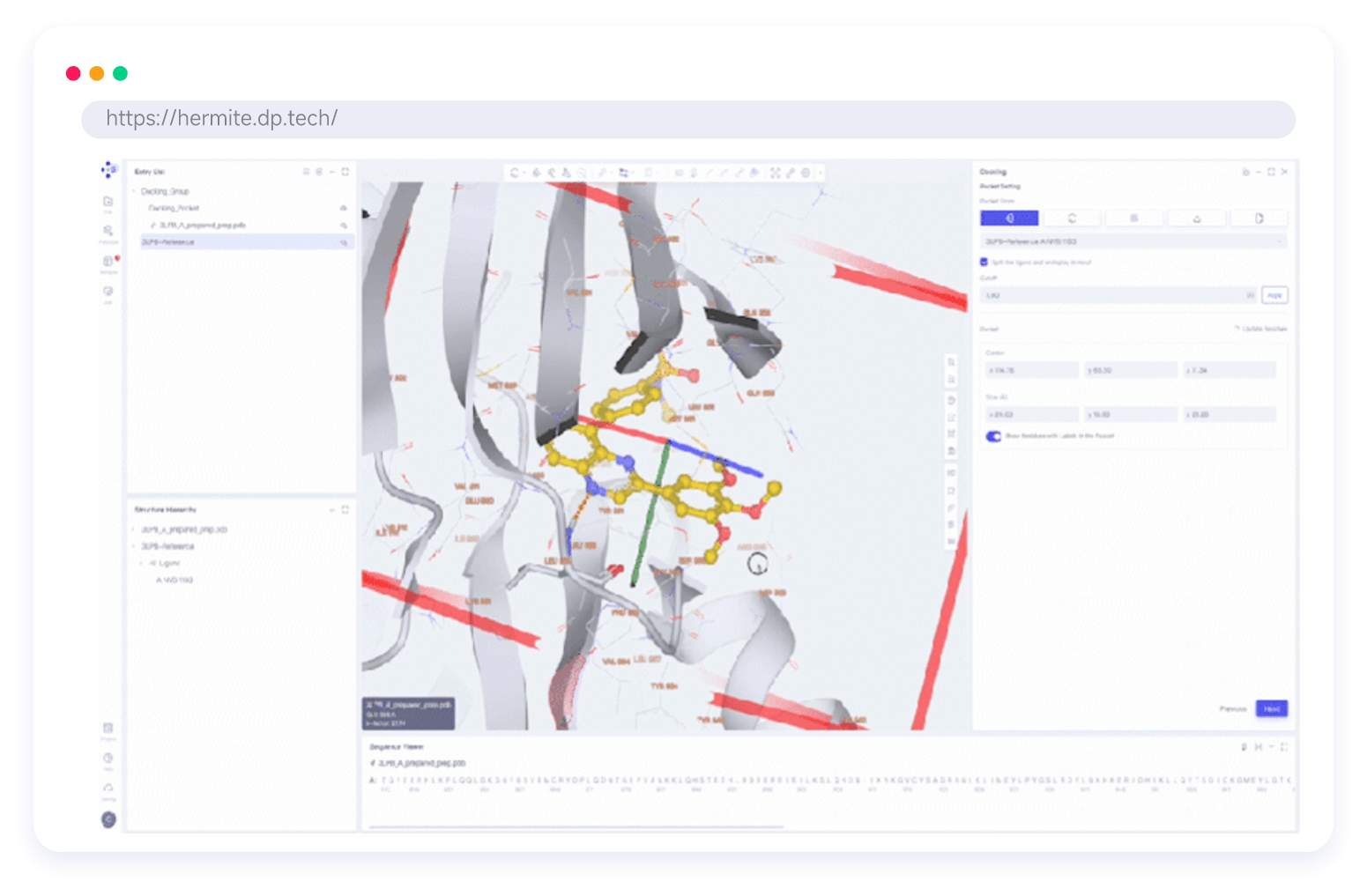
Linked 2D & 3D structure visualization and editing, supporting freeform design and precise calculations.
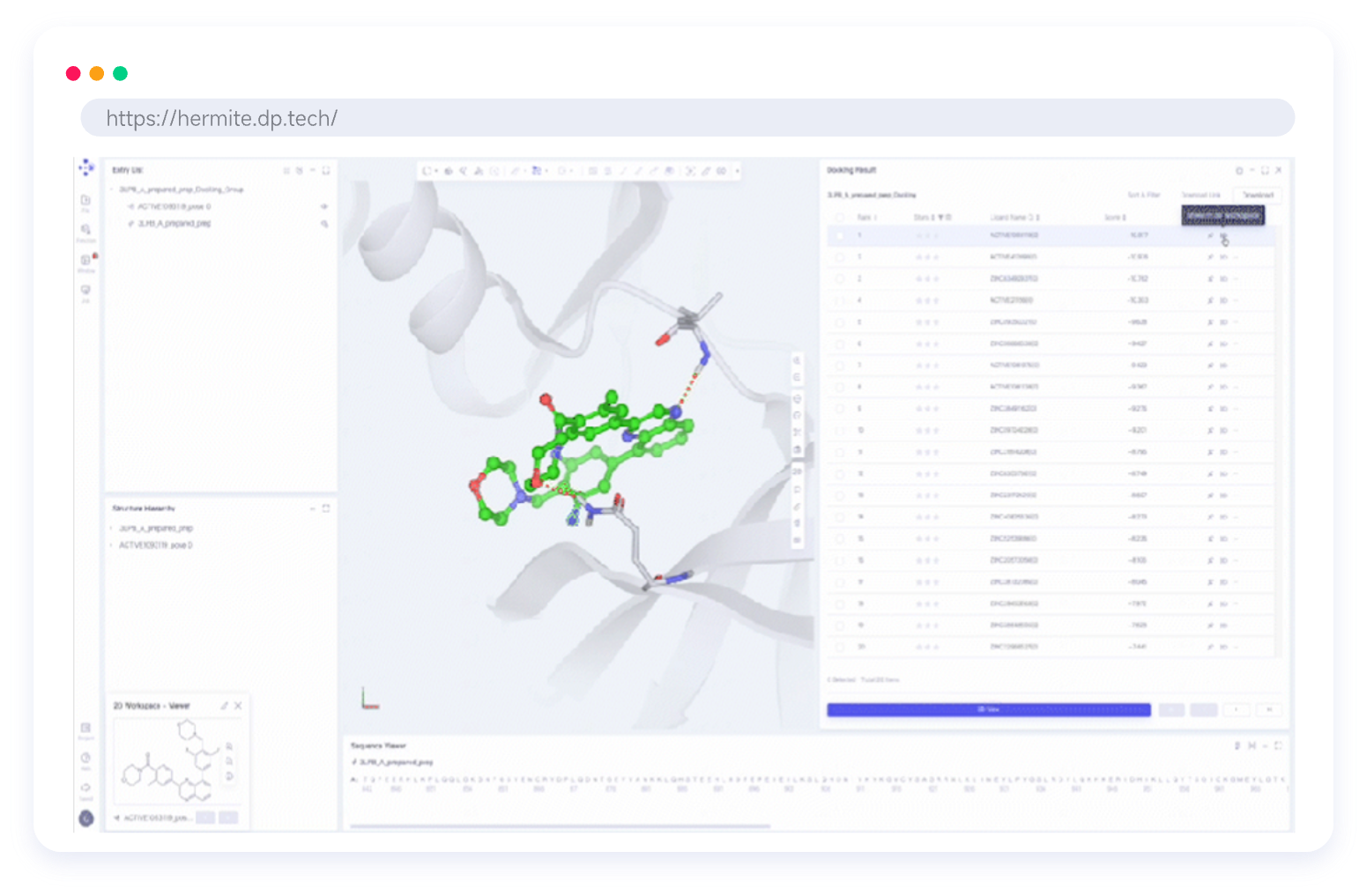
Multi-layer information display of structures, labels, and interactions, aiding in the construction of SAR models for informed decision-making
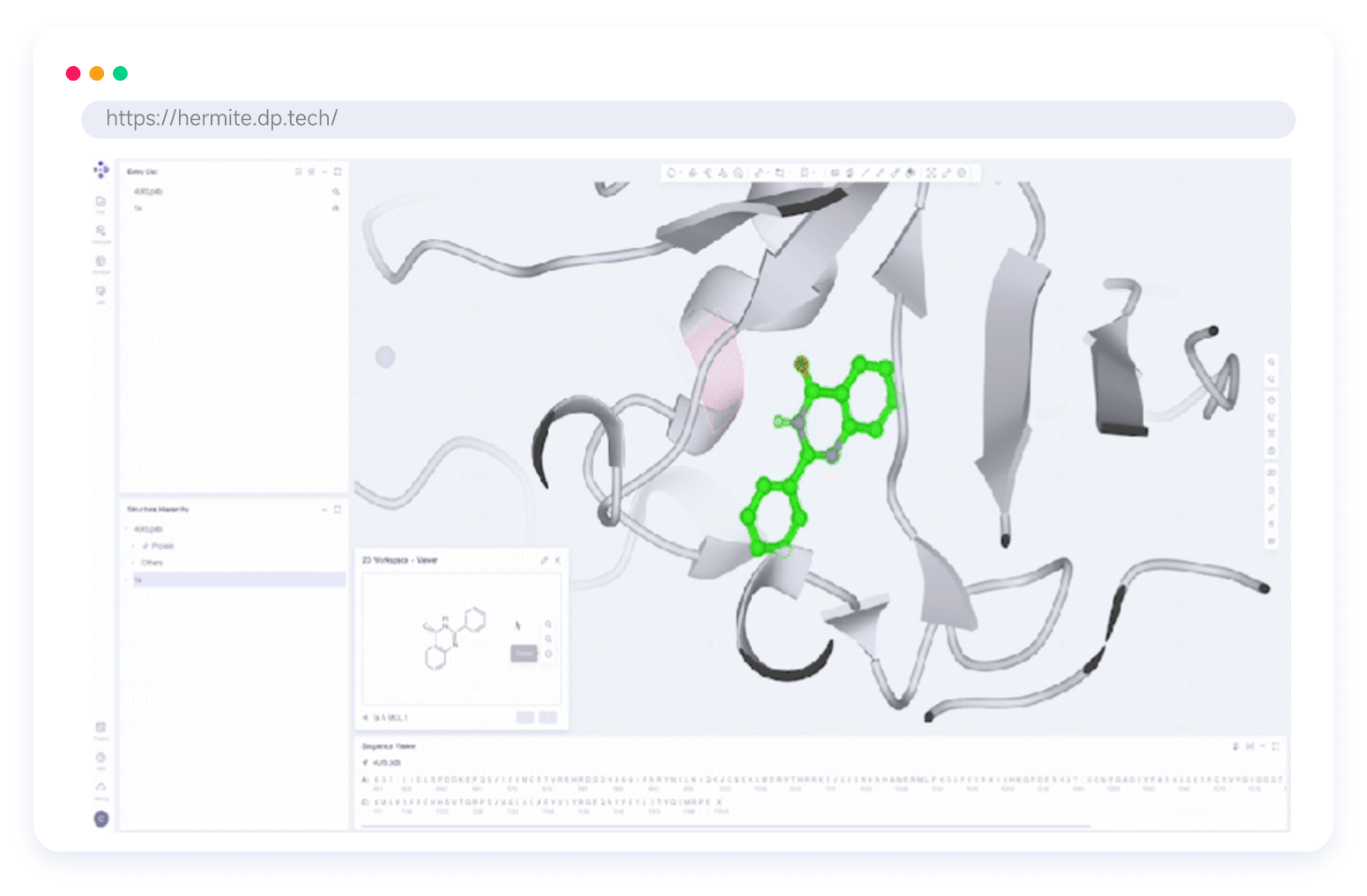
Millisecond-level molecular structure switching, providing a smooth experience directly in the browser.
Linked 2D & 3D structure visualization and editing, supporting freeform design and precise calculations.
Multi-layer information display of structures, labels, and interactions, aiding in the construction of SAR models for informed decision-making
Millisecond-level molecular structure switching, providing a smooth experience directly in the browser.
Linked 2D & 3D structure visualization and editing, supporting freeform design and precise calculations.
.png)

Security Qualifications
Certified with SOC 2 Type II, ISO 20000, ISO 27001, and ISO 9001.

System Protection
Equipped with WAF and advanced protection measures from leading cloud providers, featuring comprehensive identity verification and access control.

Dual-Insurance Mechanism
Utilizes a dual-token mechanism for data transmission and storage, ensuring robust security for user data.
Hermite® Trusted by Customers, Accelerating Pipeline Development
- Over 60% of leading pharmaceutical companies in China have chosen the Hermite® platform.
- Customers have completed more than 200,000 Hermite® Uni-FEP calculation tasks.
- The Hermite® platform has been applied in over 50 drug pipeline projects.



Partners


























































